Protein-Protein Interaction (BioCreAtIvE2 Challenge)
The Tsujii-lab group participated in the 2006 Second BioCreative text mining challenge(http://biocreative.sourceforge.net/biocreative_2.html). We participated in the Protein-Protein Interaction Pairs subtask (BC2 PPI IPS), and our Team ID was T19 BC2 PPI. This subsection is based on work done by Sætre et al. [7].
1. Introduction
The goal of this work was to evaluate how well the AKANE system could perform on a full-text Protein-Protein Interaction (PPI) Information Extraction (IE) task. AKANE is a recently developed, sentence-level PPI system that achieved a 57.3 F-score on the AImed corpus [8]. In order to use the AKANE system for the BioCreative task, the given training data had to be preprocessed. The BioCreative training data consisted of a list of interacting protein pair identifiers for each given full-text article, while the expected input for the AKANE system is annotated sentences, as in the AImed corpus. In order to transform the full-text articles into AImed sentence-level annotations, the text was first stripped of all HTML coding to get a plain text representation. Then, each mention of protein names were tagged by a Named Entity Recognizer (NER), and all interacting and co-occurring pairs in single sentences were used for training. A Pipeline architecture was chosen to deal with each of these challenges separately. Some post-processing was also necessary, in order to transform the results from the AKANE system into the expected format for the BioCreative2 challenge. The post-processing included filtering and ranking the results, and balancing precision and recall to maximize the F-score.
2. Content
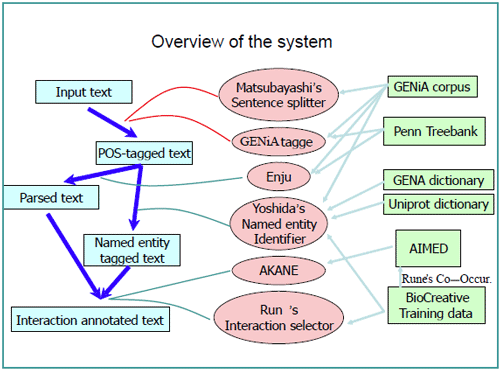
Our system was implemented using a pipeline architecture, where separate modules were put together to deal with each of the following tasks: Sentence Detection (SD), Named Entity Recognition (NER), Parsing, and Protein-Protein-Interaction (PPI) extraction. All the modules use some form of machine learning to maximize the performance on small manually annotated biological training corpora. A separate module was also made for transforming the article-level BioCreative training data into a sentence level AImed PPI-style format. Figure 1 shows an overview of the pipeline and each of the modules are briefly described in the following subparagraphs.
3. Sentence Detection
The sentence splitter for biomedical text was trained by a maximum entropy (MaxEnt) method [1], and it employs the GENiA corpus for training [4]. First, the sentence splitter detects candidate positions for splitting using selected delimiters: periods, commas, single/double quotation marks, right parentheses, etc. Then, it classifies whether the positions really split sentences or not. Features used by the classifier are: Delimiters, Previous/Next words, and information about special characters, numbers and capitalization. Some transformations of the words, like removing commas, parentheses, etc. and making lower-case versions were also used. The classifier achieved an F-score of 99.7 on 200 unseen GENiA abstracts. However, there seem to be slightly more errors on the BioCreative data set, mainly because of full text HTML encoding and figure explanation texts.
4. Named Entity Recognition
The named entity identifier takes sentence-split, POS-tagged sentences as input. It first applies a statistical named entity recognizer to the input. The statistical recognizer was trained on the data provided by the JNLPBA [5] shared task for named entity recognition. The named entity recognizer outputs marginal distributions of the probability that a substring of the sentence is a protein name. The substrings that have probabilities above some threshold are taken as protein candidates. Then such candidates are mapped to dictionary items whose string edit distance from the candidates are less than some threshold. IDs were taken from Uniprot augmented with the GENA dictionary [6]. When a name is ambiguous, a MaxEnt classifier is used to rank the candidate IDs. The classifier is trained on 296 articles from the training data, using the following features: Similarity between the target article and the MEDLINE articles which is referred to by the Uniprot entry; Similarity between the target article and the MEDLINE articles which include the organism name which is specified by the Uniprot entry; Source dictionary (Uniprot/GENA); Edit distance of the dictionary item and the target string; and Type of the dictionary item (e.g. protein name, gene name, etc.). Similarity was estimated by the cosine measure of the articles represented by tf-idf vectors. Probabilities assigned to each ID by the MaxEnt classifier are output and used by the filtering module (described below).
5. AKANE System
For protein pair extraction, the AKANE system [8] was used. It requires AImed Corpus style [2] input for training, so a preprocessor was made to automatically create this kind of co-occurrence sentence collection for the interacting proteins. The AKANE system parses the input text using the Enju HPSG parser for bio-English. Although the parser has been trained with newswire articles, i.e. Penn Treebank, it can compute accurate analyses of biomedical texts owing to our method for domain adaptation, using the GENiA Treebank [4] to adjust the parsing model. The evaluated bio-performance is 86.9 F-score [3]. The AKANE system combines the output from the parser with the protein pair info from NER, to create the smallest connected parse tree (raw pattern) that covers both proteins. Extra new patterns (combination patterns) are also generated by recombining the parts of the raw patterns. Then, counting is done on the training corpus, to evaluate how accurate the patterns are in predicting (only) true interactions. The output from AKANE system lists all possible interactions, so for one mention of an interaction in the text, several interactions are suggested. This is because each protein name is usually ambiguous among several candidate protein IDs, so a postprocessor was made to pick (only) the most likely interaction pair, based on NER probabilities. This step is explained in detail at the end of the following subsection about pre- and post processing.
6. Training Data Generation
All sentences containing two or more proteins from the PPIs given in the training data files from BioCreative were extracted, and transformed into an AImed style XML marked-up corpus that could be used to train the AKANE system. We assumed that all sentences with a co-occurrence of two interacting (according to the training data from BioCreative) proteins really were describing that interaction. The accuracy of this assumption, and the effect it had on the prediction phase, was not properly measured (due to lack of time), but some manual inspection of the created corpus indicated reasonable accuracy. In addition, only 250 of the total 740 training articles could be used for training. The reason for this is that the AKANE system did not scale well to the large amount of text, compared to the much smaller AImed corpus. We decided to use only the co-occurrence sentences, and only from the articles where all interacting protein names/IDs could be recognized by NER. Some articles with too many co-occurrence sentences were also dropped, because of the scalability constraints.
7. Pair Filtering based on NER scores
In order to deal with ambiguity, only the single most likely protein ID were picked from any fragment of ambiguous text, and only the 20 most likely PPI pairs (based on multiplying the NER probabilities) for each article were reported. In run numbers 2 and 3, a filter was made to remove all pairs that did not have identical species tags in the last part of their protein identifiers. For example, a suggested interaction between P19235 (epor human) and Q62225 (cish mouse) is automatically filtered away, because there are not many interactions between proteins in different species. However this filter is not perfect, since there is a physical interaction reported in article PMID 11781573 between these two identifiers.
8. Results and Discussion
16 groups participated in this BioCreative task, and 45 runs were submitted. Table 1 shows the F-score, precision and recall values for our 3 runs, along with the preliminary average results provided by the BioCreative organizers. The three runs were made as follows: Run1 is a version of the system not using the inter-species interaction filter. It achieved an overall F-score of 10.5 (P: 8.2% and R: 14.6%). Run2 was the best run in terms of F-score based on the training set. On the test data it achieved an overall F-score of 13.7 (P: 10.6% and R: 19.1%). Run3 was the original AKANE system, trained with the AImed corpus, and optimized for best F-score on the training set. We did not have time to use the machine learning component of AKANE system (F-score 57.3), so instead we used manually tuned parameters and a threshold value reported to achieve 42.0 F-score on the AImed corpus (P:70% and R:30%). Still, in the evaluation, Run3 performed best, with an overall F-score of 15.8 (P: 15.7% and R: 15.9%). This means that training on full text co-occurrence training sentences did not perform any better than training on AImed abstracts alone. The reason for this is that the automatic generation of the training corpus included some noise, in terms of “interacting” co-occurrence, for example: “A and B were bought from Santa Cruz inc.”
9. Conclusions and Future Directions
Run3 gave the highest performance for our system. This suggests that we have to find a better way for training than simple co-occurrence. This in turn means that we need more annotated training corpora. Some work in this direction has already begun in the lab. (See the subsection about GENiA).
Name/Value |
Run1 |
Run2 |
Run3 |
Average all groups |
Top 3 groups |
|---|---|---|---|---|---|
F-score |
10.5 |
13.7 |
15.8 |
08.4 |
14.0 |
Precision |
08.2% |
10.6% |
15.7% |
10.2% |
19.5% |
Recall |
14.6% |
19.1% |
15.9% |
11.5% |
19.1% |
External References
- Berger A.L., Pietra S.D., and Pietra V.J.D., A maximum entropy approach to natural language processing, Computational Linguistics, 22(1):39-71, 1996. (URL: http://citeseer.ist.psu.edu/berger96maximum.html)
- Bunescu R.C. and Mooney R.J., Subsequence kernels for relation extraction, in NIPS, 2005.
- Koike A. and Takagi T., Gene/protein/family name recognition in biomedical literature, in Proc. Biolink 2004, 9-16, 2004.
Internal Publications
- Hara T., Miyao Y., and Tsujii J., Adapting a probabilistic disambiguation model of an HPSG parser to a new domain, in R. Dale, K.F. Wong, J. Su, and O.Y. Kwong, eds., IJCNLP 2005 , volume 3651 of LNAI , 199-210, Springer-Verlag, Jeju Island, Korea, October 2005, ISSN 0302- 9743.
- Kim J.D., Ohta T., Tateishi Y., and Tsujii J., GENiA corpus - a semantically annotated corpus for bio-textmining, Bioinformatics, 19(suppl. 1):i180-i182, 2003, ISSN 1367-4803.
- Kim J.D., Ohta T., Tsuruoka Y., Tateishi Y., and Collier N., Introduction to the bio-entity recognition task at JNLPBA, in Proceedings of the JNLPBA-04, 70-75, Geneva, Switzerland, 2004.
- Sætre R., Yoshida K., Yakushiji A., Miyao Y., Matsubayashi Y., and Ohta T., Akane system: Protein-protein interaction pairs in biocreative2 challenge, PPI-IPS subtask, in Proceedings of the Second BioCreative Challenge Evaluation Workshop, CNIO, Spain, April 2007, ISBN 84-933255-6-2.
- Yakushiji A., Relation Information Extraction Using Deep Syntactic Analysis, Ph.D. thesis, University of Tokyo, 2006.